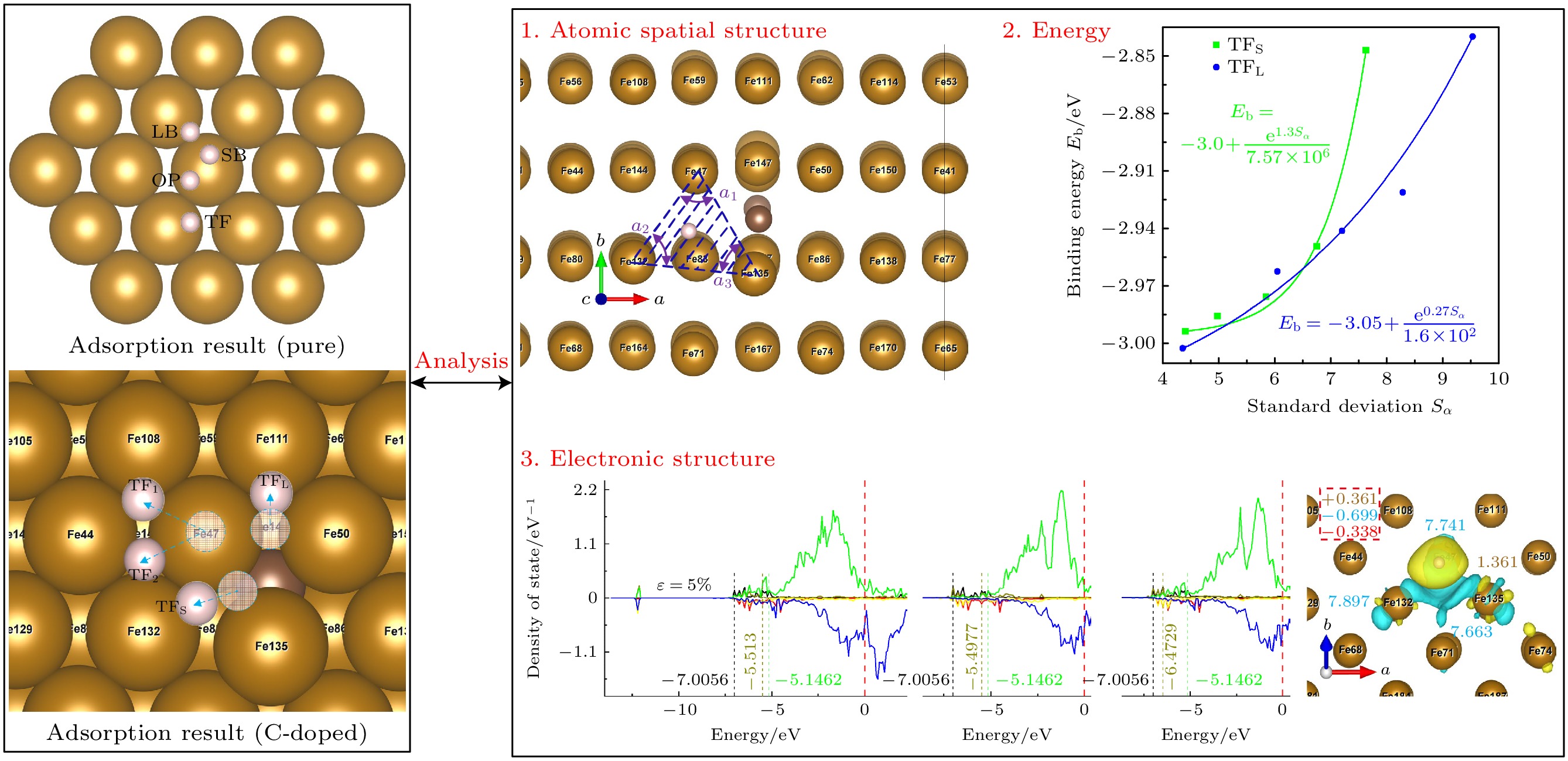
In order to further investigate and improve the mechanism of the interaction between pre-strain/pre-stress and hydrogen-adsorbed steel (Fe-C alloy) surface at the microstructural level, the first-principles calculations method is used to study the effects of uniaxial pre-strain on hydrogen adsorption and diffusion on C-doped Fe(110) surface. The influence of pre-strain on hydrogen adsorption and permeation is investigated from three aspects: surface atomic spatial configuration, binding energy (Eb), and electronic structure. The diffusion energy barriers for hydrogen permeation are calculated in both doped and undoped C atoms. The results demonstrate that doped C atoms induce octahedral lattice distortion in Fe crystals in different directions, creating “distortion” on the Fe(110) surface. Variations in distortion degree (DΔ) at different sites and their distances from C atom lead to inconsistent trends in adsorption configurations (H adsorption height d and unit surface area SΔ) and binding energy (Eb) under pre-strain. For adsorption configurations, d is coupled by ε and C atom effects: at the TFpure site (non-C-doped site ), d decreases as SΔ increases; under compression (ε decreases from 0% to –5%) at TF (C-doped site with C atom directly beneath the site), TFS (C-doped site located closer to the maximally distorted atom Fe135) and TFL sites (C-doped site located farther from the maximally distorted atom Fe135), d positively correlates with DΔ, while under tension (ε increases from 0% to 5%), d negatively correlates with SΔ. As ε increases from –5% to 5%, Eb peaks at TFpure then declines, whereas Eb at TF decreases initially before rising, and Eb at TFS/TFL monotonically increases. The analysis hows that Eb at TFS/TFL positively is correlated with the standard deviation (Sα) of the three internal angles in the triangular unit. The trend of diffusion energy barrier (E∆) is opposite to that of Eb. When H is adsorbed at C-doped sites, the adsorption configuration and binding energy calculations indicate that H tends to diffuse inward more readily. However, electronic structure analysis reveals repulsion between C and H atoms, accompanied by increased diffusion barriers compared with the scenarios in the undoped cases, causing H atoms to accumulate around C atoms rather than penetrate the bulk phase, thereby leading hydrogen atoms to embrittle. The calculations of adsorption configuration, binding energy, and diffusion barrier indicate that at doped sites (TFS site), increasing tensile strain can contribute to H diffusion into the steel microstructure, whereas compressive strain hinders it. This explains the engineering phenomenon where “higher carbon content exacerbates hydrogen embrittlement tendency under equivalent stress” on an atomic scale. This work elucidates the mechanism of H adsorption on pre-strained Fe-C alloy surfaces from an electronic structure perspective, providing theoretical ideas for studying hydrogen embrittlement.











 下载: 导出CSV
下载: 导出CSV



 下载:
下载:











